Exercise I: Exploratory Data Analysis (EDA)¶
In our first exercise, we will explore a public dataset of Coronavirus PCR tests based on a fascinating blog post published as part of the MAFAT Challenge.
The purpose of this exercise is to demonstrate the importance of inspecting data and understanding it before trying to do anything fancy. As elementary as it may sound, this preliminary step is the pitfall of many data analysis pipelines and nurturing an instinct for visualizing and organizing your raw data will pay off tremendously both in the process of developing and in evaluating the robustness of your models.
We’ll start things off by summarizing some key information about the dataset:
The dataset is shared as a GitHub repository containing a directory of CSV files.
It is collected by two US-based companies providing services in the health industry.
The number of observations is still relatively small, but more are added weekly and the quality of the data is high (there are many features to work with and it is relatively organized and “clean”).
There are also chest X-rays available for some of the patients, that were not included in this analysis.
So far so good, let’s get to work.
Data Retrieval¶
In order to keep things orderly and easy to maintain, we will first create a file with some general configurations:
"""
configuration.py
Constants defined for data retrieval and preprocessing.
"""
# GitHub data directory URL.
DATA_DIRECTORY = (
"https://raw.githubusercontent.com/mdcollab/covidclinicaldata/master/data/"
)
# Pattern for composing CSV file URLS to read.
CSV_FILE_PATTERN = "{week_id}_carbonhealth_and_braidhealth.csv"
CSV_URL_PATTERN = f"{DATA_DIRECTORY}/{CSV_FILE_PATTERN}"
# *week_id*s used to format CSV_FILE_PATTERN with.
WEEK_IDS = (
"04-07",
"04-14",
"04-21",
"04-28",
"05-05",
"05-12",
"05-19",
"05-26",
"06-02",
"06-09",
"06-16",
"06-23",
"06-30",
"07-07",
"07-14",
"07-21",
"07-28",
"08-04",
"08-11",
"08-18",
"08-25",
"09-01",
"09-08",
"09-15",
"09-22",
"09-29",
"10-06",
"10-13",
"10-20",
)
# Values replacement dictionary by column name.
REPLACE_DICT = {"covid19_test_results": {"Positive": True, "Negative": False}}
# Name of the column containing the COVID-19 test results.
TARGET_COLUMN_NAME = "covid19_test_results"
# Fractional threshold of missing values in a feature.
NAN_FRACTION_THRESHOLD = 0.5
# Prefix used in columns containing X-ray data results.
X_RAY_COLUMN_PREFIX = "cxr_"
And then write a short module to create a read_data() method:
Technical Note: remove_x_ray_columns
def remove_x_ray_columns(
data: pd.DataFrame, prefix: str = X_RAY_COLUMN_PREFIX
) -> pd.DataFrame:
"""
Removes radiology results columns from the dataset.
Parameters
----------
data : pd.DataFrame
Input dataset
Returns
-------
pd.DataFrame
Dataset without X-ray data
"""
xray_columns = [
column_name
for column_name in data.columns
if column_name.startswith(prefix)
]
return data.drop(xray_columns, axis=1)
def read_data() -> pd.DataFrame:
"""
Returns the public COVID-19 PCR test dataset from the *covidclinicaldata*
repository.
Returns
-------
pd.DataFrame
COVID-19 PCR test dataset
"""
# Read CSVs from remote location as a single dataframe.
urls = [CSV_URL_PATTERN.format(week_id=week_id) for week_id in WEEK_IDS]
dataframes = [pd.read_csv(url, error_bad_lines=False) for url in urls]
data = pd.concat(dataframes, ignore_index=True)
# Convert COVID-19 test results to boolean values.
data.replace(REPLACE_DICT, inplace=True)
# Remove x-ray data.
data = remove_x_ray_columns(data)
# Remove the batch date column.
data.drop("batch_date", axis=1, inplace=True)
# Remove rows with no test results.
data.dropna(axis="rows", subset=[TARGET_COLUMN_NAME], inplace=True)
return data
Now we can read the data by simply running:
data = read_data()
/home/runner/work/ml_for_neuro/ml_for_neuro/source/chapters/chapter_01/read_data.py:57: FutureWarning: The error_bad_lines argument has been deprecated and will be removed in a future version.
dataframes = [pd.read_csv(url, error_bad_lines=False) for url in urls]
Raw Inspection¶
Technical Note: get_scattered_chunks
"""
get_scattered_chunks.py
Definition of the `get_scattered_chunks()` function, used to generate
a subsample of rows for manual review of the dataset.
"""
import numpy as np
import pandas as pd
def get_scattered_chunks(
data: pd.DataFrame, n_chunks: int = 5, chunk_size: int = 3
) -> pd.DataFrame:
"""
Returns a subsample of equally scattered chunks of rows.
Parameters
----------
data : pd.DataFrame
Input dataset
n_chunks : int
Number of chunks to collect for the subsample
chunk_size : int
Number of rows to include in each chunk
Returns
-------
pd.DataFrame
Subsample data
"""
endpoint = len(data) - chunk_size
sample_indices = np.linspace(0, endpoint, n_chunks, dtype=int)
sample_indices = [
index for i in sample_indices for index in range(i, i + chunk_size)
]
return data.iloc[sample_indices, :]
Technical Note: print_table
We will prepare a function to make the styling of the printed dataframe better suited for our purposes:
"""
print_table.py
Defines a set of styling parameters and functions for the dataset's
table display.
"""
import math
import pandas as pd
from configuration import TARGET_COLUMN_NAME
from typing import Any
def highlight_nan(value: Any) -> str:
"""
Highlight NaN values in grey.
Parameters
----------
value : Any
Cell value
Returns
-------
str
Cell background color definition
"""
try:
value = float(value)
except ValueError:
color = "white"
else:
color = "grey" if math.isnan(value) else "white"
finally:
return f"background-color: {color}"
def highlight_positives(test_result: bool) -> str:
"""
Highlight positive values in red.
Parameters
----------
test_result : bool
Observed test result
Returns
-------
str
Cell background color definition
"""
color = "red" if test_result else "white"
return f"background-color: {color}"
def get_table_styles(
header_font_size: int = 12, cell_font_size: int = 11
) -> list:
"""
Creates a table styles definition to be used by the
`set_table_styles()` method.
References
----------
* Pandas' table styles documentation:
https://pandas.pydata.org/pandas-docs/stable/user_guide/style.html#Table-styles
Parameters
----------
header_font_size : int
Header text font size in pixels (px)
cell_font_size : int
Cell text font size in pixels (px)
Returns
-------
list
Table styles definition
"""
heading_properties = [("font-size", f"{header_font_size}px")]
cell_properties = [("font-size", f"{cell_font_size}px")]
return [
dict(selector="th", props=heading_properties),
dict(selector="td", props=cell_properties),
]
def print_table(
data: pd.DataFrame,
header_font_size: int = 12,
cell_font_size: int = 11,
text_align: str = "center",
) -> pd.DataFrame.style:
"""
Returns a styled representation of the dataframe.
Parameters
----------
data : pd.DataFrame
Input data
header_font_size : int
Header text font size in pixels (px)
cell_font_size : int
Cell text font size in pixels (px)
text_align : str
Any of the CSS text-align property options, defaults to "center"
Returns
-------
pd.DataFrame.style
Styled dataframe representation
"""
table_styles = get_table_styles(
header_font_size=header_font_size, cell_font_size=cell_font_size
)
return (
data.style.set_table_styles(table_styles)
.applymap(highlight_nan)
.applymap(highlight_positives, subset=[TARGET_COLUMN_NAME])
.set_properties(**{"text-align": text_align})
)
Tip
For more information about styling Pandas dataframes, see the documentation.
data_chunks = get_scattered_chunks(data, n_chunks=5, chunk_size=3)
print_table(data_chunks)
| test_name | swab_type | covid19_test_results | age | high_risk_exposure_occupation | high_risk_interactions | diabetes | chd | htn | cancer | asthma | copd | autoimmune_dis | smoker | temperature | pulse | sys | dia | rr | sats | rapid_flu_results | rapid_strep_results | ctab | labored_respiration | rhonchi | wheezes | days_since_symptom_onset | cough | cough_severity | fever | sob | sob_severity | diarrhea | fatigue | headache | loss_of_smell | loss_of_taste | runny_nose | muscle_sore | sore_throat | er_referral | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | SARS COV 2 RNA RTPCR | Nasopharyngeal | False | 58 | True | nan | False | False | False | False | False | False | False | False | 36.950000 | 81.000000 | 126.000000 | 82.000000 | 18.000000 | 97.000000 | nan | nan | False | False | False | False | 28.000000 | True | Severe | nan | False | nan | False | False | False | False | False | False | False | False | False |
| 1 | SARS-CoV-2, NAA | Oropharyngeal | False | 35 | False | nan | False | False | False | False | False | False | False | False | 36.750000 | 77.000000 | 131.000000 | 86.000000 | 16.000000 | 98.000000 | nan | nan | False | False | False | False | nan | True | Mild | False | False | nan | False | False | False | False | False | False | False | False | False |
| 2 | SARS CoV w/CoV 2 RNA | Oropharyngeal | False | 12 | nan | nan | False | False | False | False | False | False | False | False | 36.950000 | 74.000000 | 122.000000 | 73.000000 | 17.000000 | 98.000000 | nan | nan | nan | nan | nan | nan | nan | False | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | False |
| 23498 | SARS-CoV-2, NAA | Nasal | False | 35 | False | False | False | False | False | False | False | False | False | False | 37.000000 | 69.000000 | 136.000000 | 84.000000 | 12.000000 | 100.000000 | nan | nan | False | nan | nan | nan | nan | False | nan | False | False | nan | False | False | False | False | False | False | False | False | nan |
| 23499 | SARS-CoV-2, NAA | Nasal | False | 24 | False | True | False | False | False | False | False | False | False | False | 36.750000 | 70.000000 | 128.000000 | 78.000000 | 12.000000 | 99.000000 | nan | nan | nan | False | False | False | nan | False | nan | False | False | nan | False | False | False | False | False | False | False | False | nan |
| 23500 | SARS-CoV-2, NAA | Nasal | False | 52 | False | False | False | False | False | False | False | False | False | False | 37.000000 | 94.000000 | 165.000000 | 82.000000 | 12.000000 | 98.000000 | nan | nan | nan | False | False | False | 7.000000 | True | nan | False | False | nan | False | False | False | False | False | False | False | False | nan |
| 46996 | SARS-CoV-2, NAA | Nasal | False | 11 | False | False | False | False | False | False | False | False | False | False | 36.900000 | 78.000000 | 116.000000 | 79.000000 | 16.000000 | 100.000000 | nan | nan | True | False | True | True | nan | False | nan | False | False | nan | False | False | False | False | False | False | False | False | nan |
| 46997 | SARS-CoV-2, NAA | Nasal | False | 30 | False | False | False | False | False | False | False | False | False | False | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | False | nan | False | False | nan | False | False | False | False | False | False | False | False | nan |
| 46998 | SARS-CoV-2, NAA | Nasal | False | 36 | False | False | False | False | False | False | False | False | False | False | 36.850000 | 81.000000 | 122.000000 | 81.000000 | 14.000000 | 100.000000 | nan | nan | nan | False | nan | nan | 7.000000 | True | Mild | False | True | Mild | True | True | True | False | False | False | True | False | nan |
| 70494 | Rapid COVID-19 PCR Test | Nasal | False | 32 | False | nan | False | False | False | False | False | False | False | False | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | False | nan | nan | False | nan | False | False | False | False | False | False | False | False | nan |
| 70495 | SARS-CoV-2, NAA | Nasal | False | 59 | False | False | False | False | True | False | False | False | False | False | 36.600000 | 75.000000 | 137.000000 | 99.000000 | nan | 97.000000 | nan | nan | False | False | False | False | 7.000000 | True | Moderate | True | False | nan | False | False | False | False | False | False | False | True | nan |
| 70496 | SARS-CoV-2, NAA | Nasal | False | 27 | False | True | False | False | False | False | False | False | False | False | 37.000000 | 63.000000 | 113.000000 | 71.000000 | 15.000000 | 98.000000 | nan | nan | nan | False | nan | nan | 2.000000 | False | nan | False | False | nan | False | False | False | False | False | False | False | False | nan |
| 93992 | SARS-CoV-2, NAA | Nasal | False | 33 | False | nan | False | False | False | False | False | False | False | False | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | False | nan | nan | False | nan | False | False | False | False | False | False | False | False | nan |
| 93993 | Rapid COVID-19 PCR Test | Nasal | False | 46 | False | False | False | False | False | False | False | False | False | False | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | False | nan | False | False | nan | False | False | False | False | False | False | False | False | nan |
| 93994 | Rapid COVID-19 PCR Test | Nasal | False | 53 | False | nan | False | False | True | False | False | False | False | False | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | nan | False | nan | nan | False | nan | False | False | False | False | False | False | False | False | nan |
Alright! Some things that we can already learn about our dataset from this table are:
It contains a total of
93995observations.There are
41columns with mixed data types (nemeric and categorical).Missing values certainly exist (we can easily spot
nanentries).The subsample raises a strong suspicion that dataset is imbalanced, i.e. when examining our target variable (
'covid19_test_results') it seems there are far more negative observations than positive ones. A quicksum()call reveals that indeed only1313of the93995observations are positive.
There are at least two more built-in methods we could use to get a better sense of the information contained within each column are. The first is:
data.info()
<class 'pandas.core.frame.DataFrame'>
Int64Index: 93995 entries, 0 to 93994
Data columns (total 41 columns):
# Column Non-Null Count Dtype
--- ------ -------------- -----
0 test_name 93995 non-null object
1 swab_type 93995 non-null object
2 covid19_test_results 93995 non-null bool
3 age 93995 non-null int64
4 high_risk_exposure_occupation 93826 non-null object
5 high_risk_interactions 69168 non-null object
6 diabetes 93995 non-null bool
7 chd 93995 non-null bool
8 htn 93995 non-null bool
9 cancer 93995 non-null bool
10 asthma 93995 non-null bool
11 copd 93995 non-null bool
12 autoimmune_dis 93995 non-null bool
13 smoker 93995 non-null bool
14 temperature 47542 non-null float64
15 pulse 48279 non-null float64
16 sys 46523 non-null float64
17 dia 46523 non-null float64
18 rr 41448 non-null float64
19 sats 47535 non-null float64
20 rapid_flu_results 254 non-null object
21 rapid_strep_results 391 non-null object
22 ctab 35467 non-null object
23 labored_respiration 48747 non-null object
24 rhonchi 23344 non-null object
25 wheezes 27488 non-null object
26 days_since_symptom_onset 15865 non-null float64
27 cough 93980 non-null object
28 cough_severity 5711 non-null object
29 fever 71074 non-null object
30 sob 93789 non-null object
31 sob_severity 2836 non-null object
32 diarrhea 93808 non-null object
33 fatigue 93819 non-null object
34 headache 93812 non-null object
35 loss_of_smell 93805 non-null object
36 loss_of_taste 93805 non-null object
37 runny_nose 93808 non-null object
38 muscle_sore 93813 non-null object
39 sore_throat 93812 non-null object
40 er_referral 11169 non-null object
dtypes: bool(9), float64(7), int64(1), object(24)
memory usage: 24.5+ MB
This simple table allows us to easily infer that:
Most (
32/40) of the collected features are categorial (their data type isobjectorbool).Some features contain relatively few non-null values.
Another useful method we can use is:
data.describe()
| age | temperature | pulse | sys | dia | rr | sats | days_since_symptom_onset | |
|---|---|---|---|---|---|---|---|---|
| count | 93995.000000 | 47542.000000 | 48279.000000 | 46523.000000 | 46523.000000 | 41448.000000 | 47535.000000 | 15865.000000 |
| mean | 39.175690 | 36.794765 | 76.933677 | 123.149045 | 78.297079 | 14.710601 | 98.238224 | 7.063914 |
| std | 15.036870 | 0.289229 | 13.238270 | 16.113033 | 9.464313 | 1.971628 | 1.423986 | 17.232417 |
| min | -3.000000 | 33.500000 | 35.000000 | 50.000000 | 15.000000 | 0.000000 | 55.000000 | 1.000000 |
| 25% | 28.000000 | 36.650000 | 68.000000 | 112.000000 | 72.000000 | 13.000000 | 97.000000 | 2.000000 |
| 50% | 37.000000 | 36.800000 | 76.000000 | 121.000000 | 78.000000 | 15.000000 | 98.000000 | 3.000000 |
| 75% | 50.000000 | 36.950000 | 85.000000 | 132.000000 | 84.000000 | 16.000000 | 99.000000 | 7.000000 |
| max | 91.000000 | 39.600000 | 160.000000 | 235.000000 | 150.000000 | 40.000000 | 100.000000 | 300.000000 |
This table summarizes the basic properties of each of the numerical columns contained in the dataset and gives a brief overview of the observed variance.
Missing Values¶
Handling missing values requires careful judgement. Possible solutions include:
Removing the entire feature (column) containing the missing values.
Removing all observations with missing values.
Imputation: “Filling in” missing values with some constant or statistic, such as the mean or the mode.
There is not one correct method - different circumstances call for different approaches, and often there is still room for debate. However, some useful guidelines are:
If a column contains a small number of observations (relative to the size of the dataset) and the dataset is rich enough and offers more features that could be expected to be informative, it might be best to remove it.
If the dataset is large and the feature in question is crucial for the purposes of our analysis, remove all observations with missing values.
Imputation might sound like a good trade-off if there is a good reason to believe some statistic may adequately approximate the missing values, but it is also the subject of many misconceptions and often used poorly.
There are also learning methods that can safely include missing values (such as decision trees). We will learn when and how these are used later in this course.
Let’s first get a better sense of what fraction of the values is missing in each of the relevant columns in our dataset:
Technical Note: calculate_nan_fractions
def calculate_nan_fractions(
data: pd.DataFrame, target_column: str = TARGET_COLUMN_NAME
) -> pd.DataFrame:
"""
Calculates the fraction of missing values within each column
in the dataset.
Parameters
----------
data : pd.DataFrame
Input dataset
target_column : str, optional
Boolean target column name, by default TARGET_COLUMN_NAME
Returns
-------
pd.DataFrame
Missing value fractions
"""
# Extract columns with null values.
nan_columns = data.columns[data.isnull().any()]
nan_data = data[nan_columns]
# Calculate fractions of null values.
nan_counts = nan_data.isnull().sum()
fraction_missing = nan_counts / len(nan_data)
positives_nan_counts = nan_data[data[target_column]].isnull().sum()
negatives_nan_counts = nan_data[~data[target_column]].isnull().sum()
fraction_missing_positives = positives_nan_counts / nan_counts
fraction_missing_negatives = negatives_nan_counts / nan_counts
# Create dataframe.
fraction_missing_df = pd.DataFrame(
{
"Total Missing": fraction_missing,
"Negatives Fraction": fraction_missing_negatives,
"Positives Fraction": fraction_missing_positives,
}
)
fraction_missing_df.index.name = "Column Name"
return fraction_missing_df.sort_values("Total Missing", ascending=False)
missing_fractions_df = calculate_nan_fractions(data)
missing_fractions_df.style.format("{:,.2%}")
| Total Missing | Negatives Fraction | Positives Fraction | |
|---|---|---|---|
| Column Name | |||
| rapid_flu_results | 99.73% | 98.62% | 1.38% |
| rapid_strep_results | 99.58% | 98.62% | 1.38% |
| sob_severity | 96.98% | 98.66% | 1.34% |
| cough_severity | 93.92% | 98.94% | 1.06% |
| er_referral | 88.12% | 98.80% | 1.20% |
| days_since_symptom_onset | 83.12% | 99.18% | 0.82% |
| rhonchi | 75.16% | 98.98% | 1.02% |
| wheezes | 70.76% | 99.06% | 0.94% |
| ctab | 62.27% | 99.27% | 0.73% |
| rr | 55.90% | 99.34% | 0.66% |
| dia | 50.50% | 99.48% | 0.52% |
| sys | 50.50% | 99.48% | 0.52% |
| sats | 49.43% | 99.49% | 0.51% |
| temperature | 49.42% | 99.48% | 0.52% |
| pulse | 48.64% | 99.49% | 0.51% |
| labored_respiration | 48.14% | 99.48% | 0.52% |
| high_risk_interactions | 26.41% | 99.05% | 0.95% |
| fever | 24.39% | 99.10% | 0.90% |
| sob | 0.22% | 97.57% | 2.43% |
| loss_of_smell | 0.20% | 97.37% | 2.63% |
| loss_of_taste | 0.20% | 97.37% | 2.63% |
| diarrhea | 0.20% | 97.33% | 2.67% |
| runny_nose | 0.20% | 97.33% | 2.67% |
| headache | 0.19% | 97.27% | 2.73% |
| sore_throat | 0.19% | 97.27% | 2.73% |
| muscle_sore | 0.19% | 97.25% | 2.75% |
| fatigue | 0.19% | 97.16% | 2.84% |
| high_risk_exposure_occupation | 0.18% | 97.04% | 2.96% |
| cough | 0.02% | 100.00% | 0.00% |
This table allows us to see which columns containt a large fraction of missing values, and also shows us that the mean relative fraction of missing values in Coronavirus-positive observations ('1.47%') isn’t far from the general fraction of positives ('1.40%'). This is a good sign, and while not really proving anything, gives us good reason to believe the the dataset does not include a “missing not at random” bias.
Now, we will create a simple function to remove all columns with more than '50.00%' of missing values as well as any remaining observations which include missing values:
Technical Note: remove_missing_data_columns
def remove_missing_data_columns(
data: pd.DataFrame,
threshold: float = NAN_FRACTION_THRESHOLD,
target_column: str = TARGET_COLUMN_NAME,
) -> pd.DataFrame:
"""
Removes columns where the fraction of missing values is greater than
*threshold*.
Parameters
----------
data : pd.DataFrame
Input data
threshold : float, optional
Fraction of missing values to use as threshold, by default
NAN_FRACTION_THRESHOLD
target_column : str, optional
Boolean target column name, by default TARGET_COLUMN_NAME
Returns
-------
pd.DataFrame
Dataframe without missing data columns
"""
missing_fractions = calculate_nan_fractions(
data, target_column=target_column
)
flagged = missing_fractions[
missing_fractions["Total Missing"] > threshold
].index
return data.drop(flagged, axis=1)
def clean_missing_values(
data: pd.DataFrame,
threshold: float = NAN_FRACTION_THRESHOLD,
target_column: str = TARGET_COLUMN_NAME,
) -> pd.DataFrame:
"""
Cleans missing values from the dataset.
Parameters
----------
data : pd.DataFrame
Input dataset
threshold : float, optional
Fraction of missing values to use as threshold for feature removal,
by default NAN_FRACTION_THRESHOLD
target_column : str, optional
Boolean target column name, by default TARGET_COLUMN_NAME
Returns
-------
pd.DataFrame
NaN-free dataset
"""
data = remove_missing_data_columns(
data, threshold=threshold, target_column=target_column
)
return data.dropna(axis=0, how="any").reset_index(drop=True)
And run it:
data = clean_missing_values(data)
Our dataset is now clean from any missing values and ready for further inspection.
X = data.drop(TARGET_COLUMN_NAME, axis=1)
categorial_features = X.select_dtypes(["object", "bool"])
numerical_featuers = X.select_dtypes(exclude=["object", "bool"])
print("Categorial features:\n" + ", ".join(categorial_features.columns))
print("\nNumerical features:\n" + ", ".join(numerical_featuers.columns))
Categorial features:
test_name, swab_type, high_risk_exposure_occupation, high_risk_interactions, diabetes, chd, htn, cancer, asthma, copd, autoimmune_dis, smoker, labored_respiration, cough, fever, sob, diarrhea, fatigue, headache, loss_of_smell, loss_of_taste, runny_nose, muscle_sore, sore_throat
Numerical features:
age, temperature, pulse, sats
We are left with 28 features and 37754 observations (718 of which are positive).
Feature Correlations¶
import matplotlib.pyplot as plt
import seaborn as sns
correlation_matrix = X.corr()
fig, ax = plt.subplots(figsize=(12, 10))
_ = sns.heatmap(correlation_matrix, annot=True)
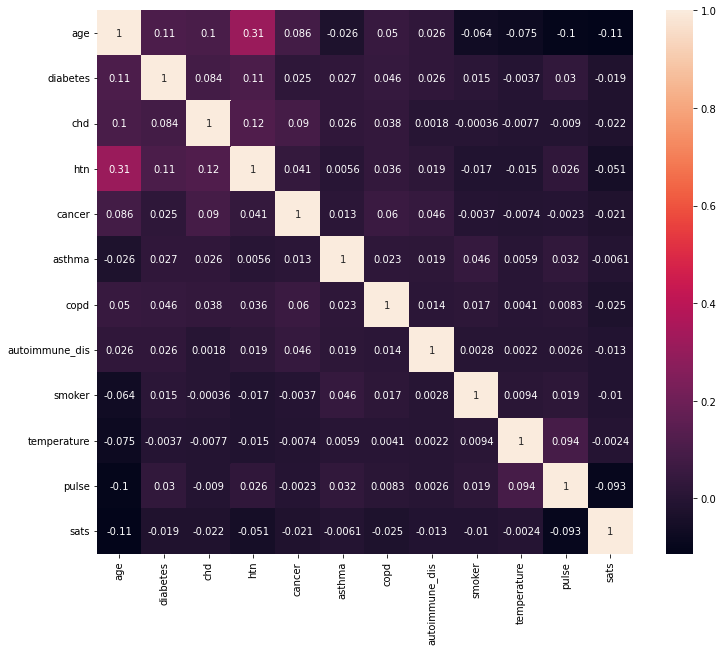
Categorial Features¶
Boolean columns previously containing missing values might have been originally inferred as object type columns, we’ll use pandas’ convert_dtypes() method to infer data types again:
X = X.convert_dtypes()
Assuming \(y\) (a COVID-19 positive patient), what is the probability of each of the features (symptomps):
import pandas as pd
boolean_features = X.select_dtypes(["bool"])
positives = boolean_features[target]
value_counts = {}
for column in positives:
value_counts[column] = positives[column].value_counts()
value_counts = pd.DataFrame(value_counts)
fig, ax = plt.subplots(figsize=(15, 5))
_ = value_counts.T.plot(kind="bar", ax=ax)
_ = plt.title("Symptoms in Positive Patients")
_ = ax.set_xlabel("Feature")
_ = ax.set_ylabel("Count")
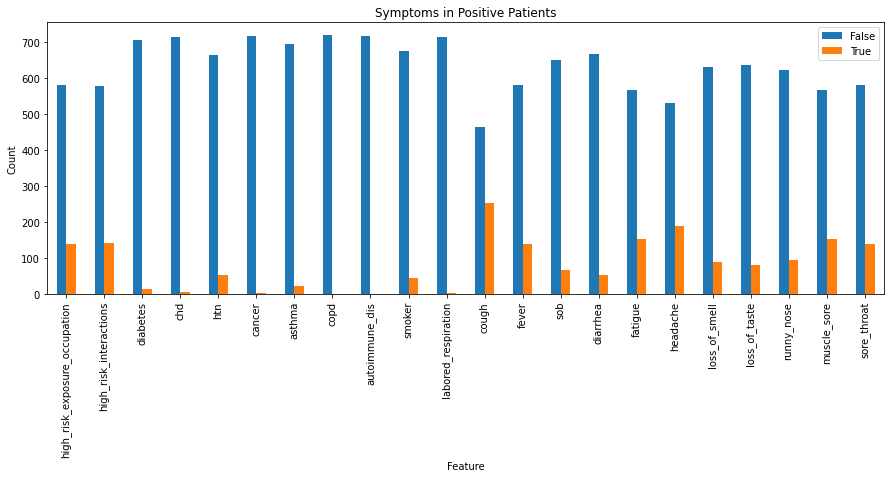
To plot the fraction of True observations in each of our target groups:
# Group all boolean features by the target variable (COVID-19 test result)
grouped = boolean_features.groupby(target)
# Calculate the fraction of positives in each group.
fractions = grouped.sum() / grouped.count()
# Create a bar plot.
fig, ax = plt.subplots(figsize=(15, 5))
_ = fractions.T.plot(kind="bar", ax=ax)
_ = plt.title("Fraction of Positive Responses by Test Result")
_ = ax.set_xlabel("Feature")
_ = ax.set_ylabel("Fraction")
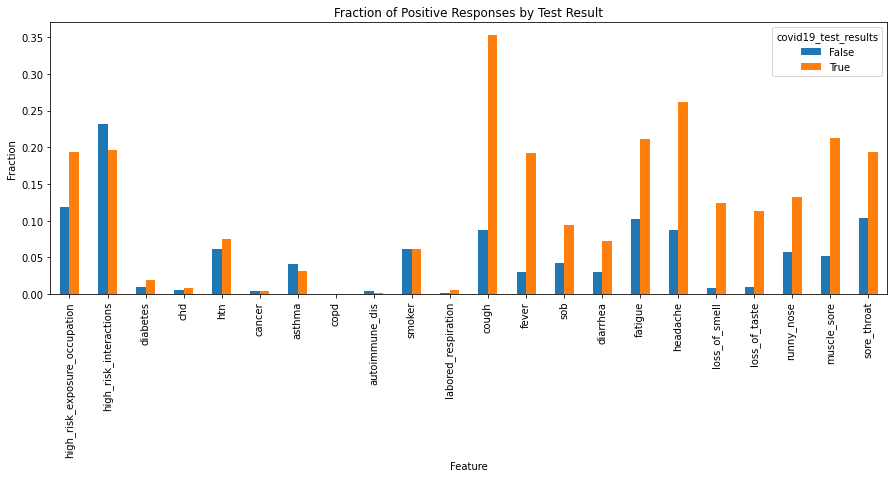
Numerical Features¶
# Select all numerical features.
numerical_features = X.select_dtypes(["float64", "int64"])
# Create distribution plots.
nrows = len(numerical_features.columns)
fig, ax = plt.subplots(nrows=nrows, ncols=2, figsize=(15, 30))
for i, feature in enumerate(numerical_features):
sns.violinplot(x=TARGET_COLUMN_NAME, y=feature, data=data, ax=ax[i, 0])
if i == 0:
ax[i, 0].set_title("Violin Plots")
ax[i, 1].set_title("Box Plots")
sns.boxplot(x=TARGET_COLUMN_NAME, y=feature, data=data, ax=ax[i, 1])
ax[i, 0].set_xlabel("")
ax[i, 1].set_xlabel("")
ax[i, 1].set_ylabel("")
_ = fig.text(0.5, 0, "COVID-19 Test Results", ha='center')
_ = fig.suptitle("Numerical Feature Distributions", y=1)
fig.tight_layout()
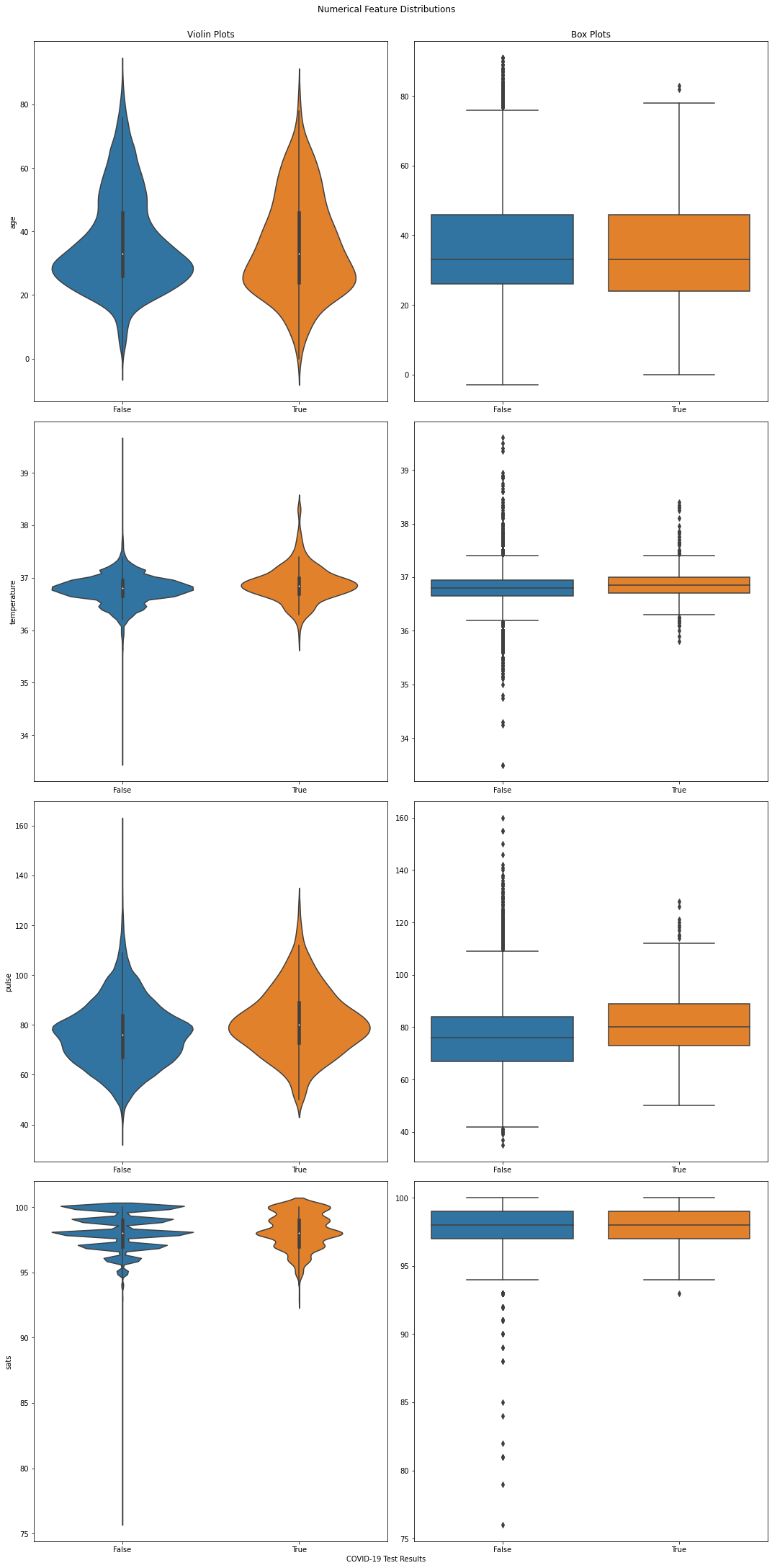
Note
Violin plots are essentially box plots combined with a kernel density estimation. While box plots give us a good understanding of the data’s quartiles and outliers, violin plots provide us with an informative representation of it’s entire distribution.